The paper describes new results in the field of algebraic biology, where matrix methods are used [Petukhov, 2008, 2012, 2013; Petuhov, He, 2010] with the transition from matrix algebra to discrete geometry and computer visualization of the genetic code. The algorithms allow to display the composition of sequences of nitrogenous bases in parametric spaces of various dimensions. Examples of visualization of the nucleotide composition of genetic sequences of various species of living organisms are given. The analysis was carried out in the spaces of binary orthogonal Walsh functions taking into account the physical and chemical parameters of the nitrogen bases. The results are compared with the rules of Erwin Chargaff concerning genetic sequences in the composition of DNA molecules. The developed method makes it possible to substantiate the relationship between DNA and RNA molecules with fractal and other geometric mosaics, reveals the orderliness and symmetries of polynucleotide chains of nitrogen bases and the noise immunity of their visual representations in the orthogonal coordinate system. The proposed methods can serve to simplify the researchers' perception of long chains of nitrogenous bases through their geometrical visualization in parametric spaces of various dimensions, and also serve as an additional criterion for classifying and identifying interspecific relationships.
DNA
and RNA nucleic acids are sequences of complementary nucleotide pairs that
perform the functions of storage and transmission of hereditary genetic
information in living organisms [1,17]. These sequences are analyzed, as a
rule, by statistical methods. They have a one-dimensional linear character and
are displayed as lines consisting of four letters of the alphabet encoding the
nucleotides: adenin (A), guanine (G), cytosine (C) and thymine (T) (uracil
(U)).
Visual
analysis of long runoffs consisting of letters encoding the nucleotides of real
genetic sequences is a laborious task. To simplify it, many algorithms and
software products have been developed that allow
to visualize
and to analyze DNA using various
histograms, tables and graphs, for example, see [23-26]. These methods are
based on machine statistical analysis and are widely used in scientific research.
In this paper we have set a task to develop a new method that simplifies the
visual analysis of long nucleotide sequences (the question of nucleotide
composition interpretation is beyond the scope of this study).
In [2] it is
shown that each nitrous base of genetic code has three variants of its binary
representation. These variants of representations, named by S.V. Cock-eared
binary sub-alphabets, differ according to the types of binary-opposition properties
in the set of nitrogenous bases:
-
G
= C "3 hydrogen bonds" / A = T "2 hydrogen bonds";
-
C
= T "pyrimidines" / A = G "purines";
-
A
= C "amino" / G = T "keto" [20];
-
A=T=G=C
(presence of phosphate residue).
Taking into
account the additional fourth feature, which is not in opposition, the system
of genetic subalphavities can be represented in the form of Hadamard's matrix
shown in Fig. 1.
Fig. 1.
A variant of the Hadamard matrix displaying the
encoding of nucleotide subalphabets. Darkened cells are +1, white cells are -1
(or vice versa depending on the encoding method). Sub-alphabet numbers are
denoted as 0, 1, 2 and 3.
This matrix is symmetric, because nucleotides can
be replaced by corresponding sub-alphabets without changing the matrix
structure (rows and columns can be changed in places) [3]. Each row and column
of the Hadamard Matrix is a Walsh function [4]. Walsh functions are a complete
set of orthogonal functions that can be used to represent any discrete function
by analogy with the use of trigonometric functions in the Fourier analysis [7].
They are used in digital engineering, in noise immunity coding, in quantum
informatics and quantum mechanics.
Chargaff revealed a system of biochemical
regularities within nucleic acid sequences, which describes quantitative
relationships between different types of nucleotides [1]. This system of
regularities is a set of algebraic relations:
1. The amount of adenin is equal to the amount
of thymine, guanine to cytosine:
A = T, G = C or A / T = 1, G / C = 1 (Watson-Crick
pairs [17]).
2. The number of purines equals the number of
pyrimidines:
A+G ≈ T+C or (A+G) / (C+T) ≈ 1.
3. number of bases with amino groups in
position 6 is equal to number of bases with ketogroups in the same position:
A+C ≈ T+G or (A+C) / (G+T) ≈ 1.
4. The ratio (A+T) / (G+C) is a specificity
factor and can be different with a predominance of AT or GC pairs depending on
a particular organism type, realizing a variety of living forms.
As can be seen from the above, the nucleotide sequence
of a living organism is a balanced system representing a double helix (DNA) and
having internal symmetries and certain mathematical regularities. Additional
information on Hadamard's symmetries and matrices in genetic coding, as well as
on genetic algebrae, is detailed in the works of S.V. Petukhov, a
biomathematician [2,10,15].
Due to the existence of a connection between
algebra and geometry (which means the existence of a connection between genetic
algebra and genetic geometries), the author has set and solved the task of developing
a method for visualizing nucleic acids. The study was based on the hypothesis
that the visualization should reflect the symmetry of the nucleotide
composition. The author's method allows investigating the phenomenon of genetic
coding from the geometric side.
The above method is an algorithm of
computer processing of biological information for scale parametric visualization
of nucleic acids in coordinate spaces of different dimensions. The main ideas
of this method were first proposed by the author in [5]. The steps of the developed
algorithm are given below.
1) Scaling. The sequence of symbols
{A,G,T,C} encoding nitrogen bases in nucleic acid is divided into fragments of
equal length N where N is a free parameter of the algorithm. The obtained
fragments of equal length will be called N-meters or N-plates [5].
2) Parametrization. Taking into account
the system of genetic subalphabets, the sequence of nitrogenous bases can be
represented as three binary sequences consisting of zeros and units. The choice
of coding method (what to consider as zero or unit) influences the rotations
and other transformations of the final visualization (therefore, for the
possibility of adequate comparison of the results obtained, it is necessary to
conduct research with reference to the "single coding standard").
3) Geometrization. Binary recording of
fragments is their representation in the form of three sequences of decimal or
other unambiguously identifying values. Converting binary N-dimensions into
decimal numbers allows them to be displayed in any coordinate system. Numerical
values specify coordinates of points in parameter space (further - in
visualization space or parametric space).
Note 1. The N-factor plays the role of
geometric visualization resolution: large N give small number of points, small
N give small coordinate grid. This fact allows us to talk about multiscale
analysis in parametric spaces.
Note 2: Steps 1 and 2 can be rearranged
(first parametrization, then scaling), which affects the computational load
when calculating long genetic sequences on a computer.
The visualization algorithm was
implemented by the author as a library of programs in Python, Lua, Moonscript
and C++ programming languages without interactive editor (GUI) and hardware
graphics acceleration. Specialized modules were used to accelerate
calculations. The average time required to process genetic information is from
several seconds to several hours depending on the scale of N and the length of
the analyzed sequence. Sometimes it was necessary to stop the counting due to exceeding
the allowed time interval. Some calculations were performed on the supercomputer
"MVS-10P" (MSC RAS).
A heuristic formula for calculating the
N scale when visualizing L-length nucleotide sequences is proposed:
,
where
square brackets are the operation of taking an integer part of a number. Width
and height of the square image in points:
It
is proposed to choose all three possible combinations of Walsh basic function
pairs as two-dimensional projection spaces. In this case, the most informative
variant of the combination of these functions may depend on a particular organism
type. At the moment it seems that there are formal rules for the choice of
basic functions, but this question needs to be further studied by analysis of the
structure of a large number of DNA of different species of organisms by the
proposed method.
The
method refers to the development of statistical methods of analysis of
nucleotide sequences and is based on parametrization, scaling and geometry of
physical and chemical parameters of the molecule. As a result of the method
application, the parametric space is given, which is finite, discrete and
three-dimensional by the number of binary-opposition features. Combinatorial
properties of this space allow to display any polynucleotides for any finite N.
Arranged numeric values on coordinate axes display physical and chemical
characteristics of N-mers, as they are clearly defined by the properties of
binary-opposition subalphabets. The method allows to visualize the nucleotide
composition in different projections, with different scales and by different subalphabites
and can be used for analysis of RNA and DNA molecules.
The
proposed method of conducting research using the developed method: the
construction of examples of visualization of long nucleotide sequences from DNA
of different organisms on the basis of the proposed method:
-
in
three-dimensional space of physico-chemical parameters, which is given by three
lines of 1, 2 and 3 of Hadamard's matrix in Fig. 1;
-
in
three-dimensional space of physico-chemical parameters, which is given by three
possible combinations of rows 1-2, 1-3 and 2-3 of Hadamard's matrix in Fig. 1;
-
in
three one-dimensional spaces of physico-chemical parameters, which are given by
three rows of 1, 2 and 3 of Hadamard's matrix in Fig. 1, considered separately
and along the whole length of the molecule, that allows to take into account
the location of N-mers in the genetic sequence;
-
the
zero (bottom) line of the Hadamard matrix in Fig. 1 is not informative, as it
does not encode the binary-opposition features, so it is not considered;
-
additionally,
according to Harmuth's theory of sequential analysis [6] it is possible to
visualize by the number of elements (zeros or units), which were found in
binary representations of N-platforms in the sequences of nitrogen bases. Due
to the fact that this method is based on the total number of some or other
parameters in the N-platform, the corresponding visualization spaces will be
called integral.
In
the course of research, visual patterns were built about a hundred genomes of
protozoa, plants, fungi, animals and viruses. In this work, genomes from the
NCBI bioinformation database [14] as well as materials kindly provided by the laboratory
of Prof. N.S. Zenkin at the Center for Bacterial Cell Biology at Newcastle
University (United Kingdom) were used for visualization.
The orthogonal basis {X, Y, Z}
selected as a three-dimensional Cartesian coordinate system gives a
visualization, an example of which is shown in Fig. 2. Each point corresponds
to the generalized characteristics of the considered binary and positional
features of the corresponding fragment of the sequence, which allows to display
the nucleotide composition of the molecule.
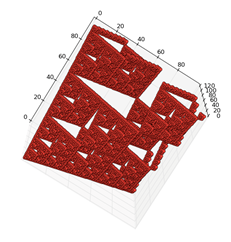
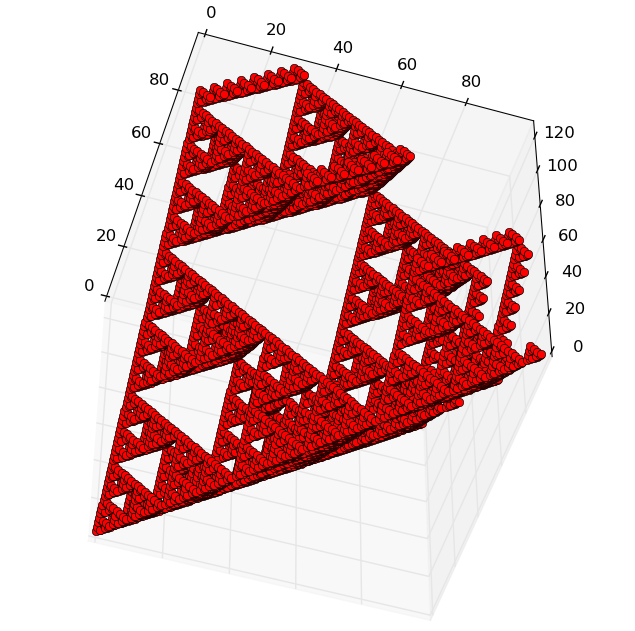
Fig.2.
Illustrations
of the three-dimensional representation of the nucleotide composition on the
example of a chromosome of a living organism in various projections,
constructed by the author's algorithm. The X, Y, and Z axes correspond to the
ascending orderly decimal representations of the binary coding of N-leafs based
on all three binary-opposition subalphabets. Each point of a figure corresponds
to N-dimensional which coordinate is set by its proton-numeric characteristics.
The analysis of three-dimensional image data is difficult because of the geometry
of the object itself. To eliminate this difficulty it is necessary to build two-dimensional
projections.
The resulting geometrical figure
resembling "Sierpinski's simplex" is typical for three-dimensional
visualization of any long nucleotide sequence. The shape of the figure is determined
by the properties of binary sub-alphabets and the Hadamard matrix in Fig. 1.
The coordinates of each point in the three-dimensional visualization space are
given by any pair of its coordinates, because the third coordinate is
calculated by adding the two remaining coordinates on the module. This algebraic
feature is associated with the redundancy of binary subalphabets used for
storing and transmitting genetic information through generations' chains. An
animated version of Fig. 2 is presented in animation:
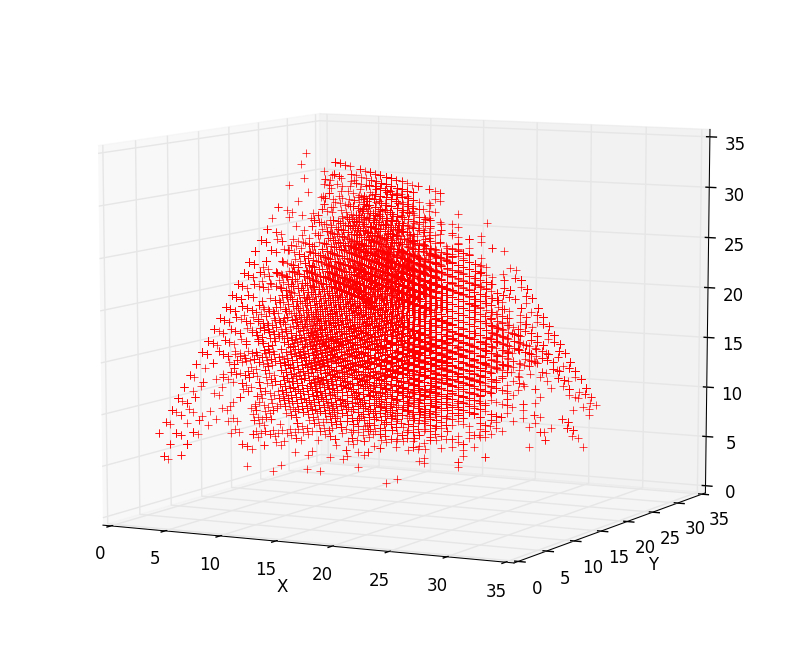
An example of the integral
three-dimensional representation of the nucleotide composition of the
chromosome of a living organism is given in Fig. 6. 3. It is an object of
finite geometry, each point of which corresponds to a set of N-dimensions of
nucleic acid, united by the number of units in binary encoding.
Fig.3.
Integral three-dimensional representation of
the nucleotide composition of the chromosome. The X, Y, and Z axes correspond
to the number of units in the decimal representation of the binary coding of
each N-platform using three binary-opposition subalphabets.
Animated version of the picture.
3 is presented in animation:
The
properties of parametric space are such that three-dimensional representations
are not convenient for perception and analysis of features of long nucleic
acids. However, two-dimensional projections of this three-dimensional representation
are suitable for displaying the specificity of their structure. In the bases
{X, Y}, (X, Z} and {Y, Z} selected as Cartesian coordinate systems,
three-dimensional visualization gives three different two-dimensional projections
based on the corresponding sub-alphabets of physical and chemical parameters of
nucleotides.
On
the basis of the developed method of visualization and computer program it was
found out that chromosomes of different kinds of organisms have individual
features of structure. Visualization of genomes of different organisms can have
a two-dimensional pattern, which is visually similar for all chromosomes and
their arbitrary fragments, as well as for the whole considered organism. Fig.
4-9 show examples of two-dimensional visualization of different nucleotide
sequences. Next to the figures in the order A, G, T, C there are pairs of Walsh
functions, which were used for coding their physico-chemical parameters (Hadamard's
matrix rows from Fig. 1).
Based
on the noted property of genetic coding (according to which the three binary-opposition
sub-alphabets are linked to each other by an addition operation on module 2) any
pair of binary representations is sufficient to determine an arbitrary nucleic
acid. Therefore, any pair of axes is sufficient for two-dimensional
visualization of the nucleotide composition. As it turned out, the question of
determining the most informative pair of coordinate axes (and, accordingly, the
parameters taken into account) depends on the type of living organism.
As a
result of the analysis, it was found that out of three variants of
two-dimensional visualization, the most informative and symmetrical mosaics are
often mosaics based on information about the external structure of the
molecule, i.e. constructed on the elements of structures encoding the features
of amino/keto and purine/pyrimidine. Such mosaics have a detailed pattern, in
which rectangular forms are usually traced (Figs. 4, 7-9). However, in some
cases, the most pronounced and symmetrical mosaics are those based on types of
hydrogen bonds representing the internal structure of the double helix DNA.
Such mosaics are usually characterized by pronounced diagonal elements of the
pattern and are found, for example, in the mitochondria DNA of the plant
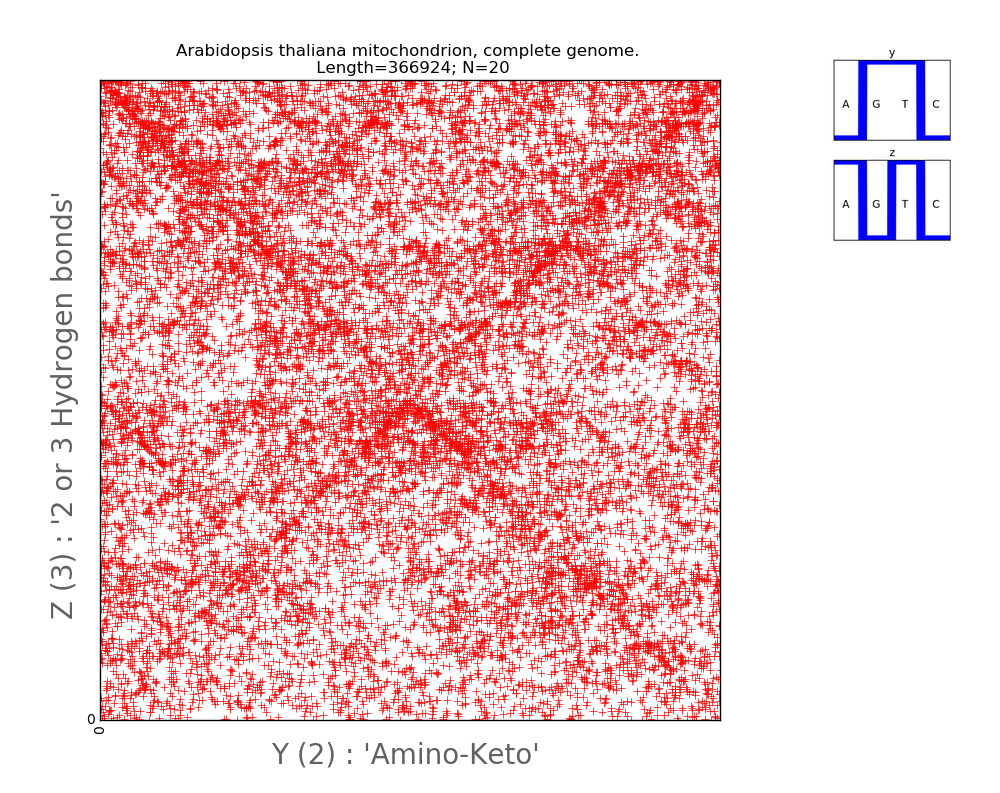
arabidopsis thaliana (Fig. 5).
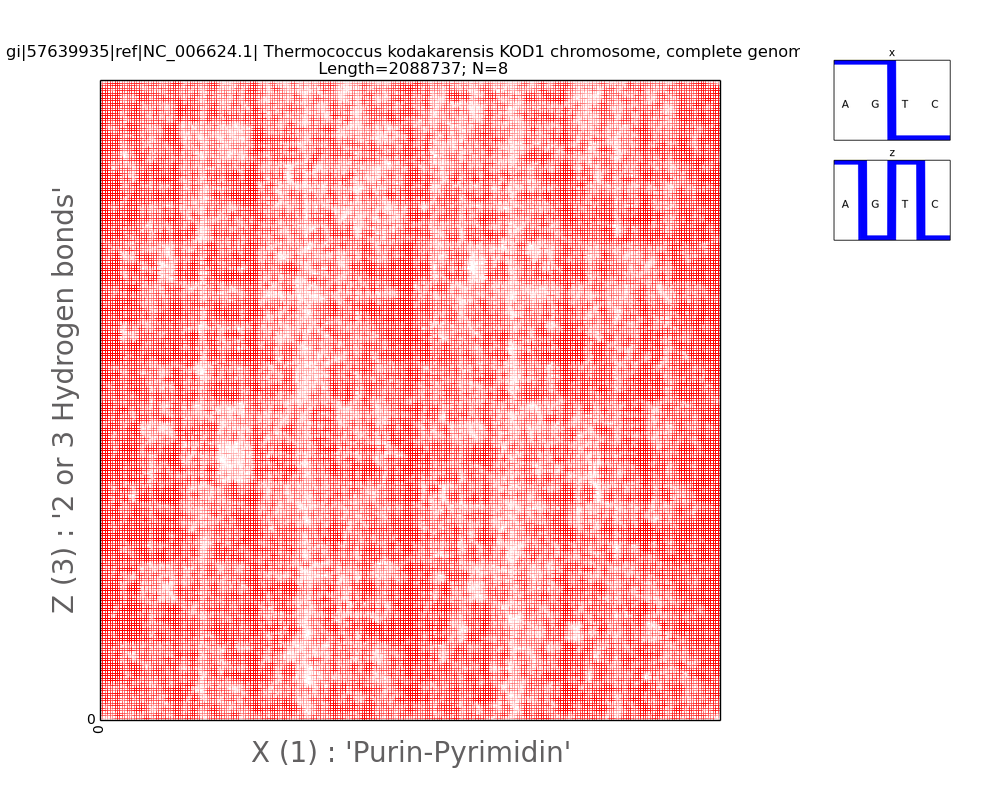
Fig.4.
Illustration
of two-dimensional representation of the nucleotide composition of the thermophilic
archaeology chromosome. A pair of Walsh functions used for parameterization is
displayed in the upper right corner. The axes of abscissa and ordinate
correspond to the decimal representation of the binary coding of each 8-wire.
Fig.5.
Illustration of two-dimensional representation of the nucleotide composition of
the mitochondria genome of the plant Rezuhovidka (Lat. Arabidopsis thaliana) of
the cabbage family (Brassicaceae). A pair of Walsh functions used for
parameterization is displayed in the upper right corner. The axes of abscissa
and ordinate correspond to decimal representations of the binary coding of each
8 weave.
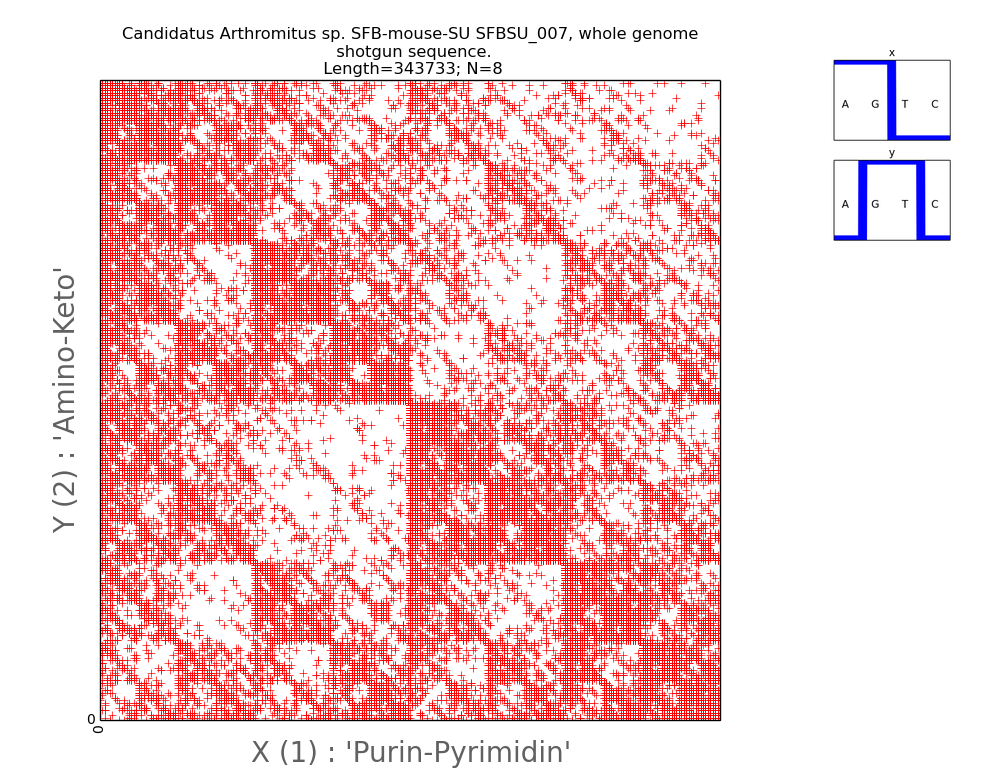
In
Fig. 6 and 7, which show a mosaic reflecting the internal structure of
chromosomes of two organisms, diagonal elements are well traced. The genome of
the bacteria in Fig. 6 shows fractal repetitions of diagonals throughout the
pattern. The diagonal elements differ in color depending on the direction and
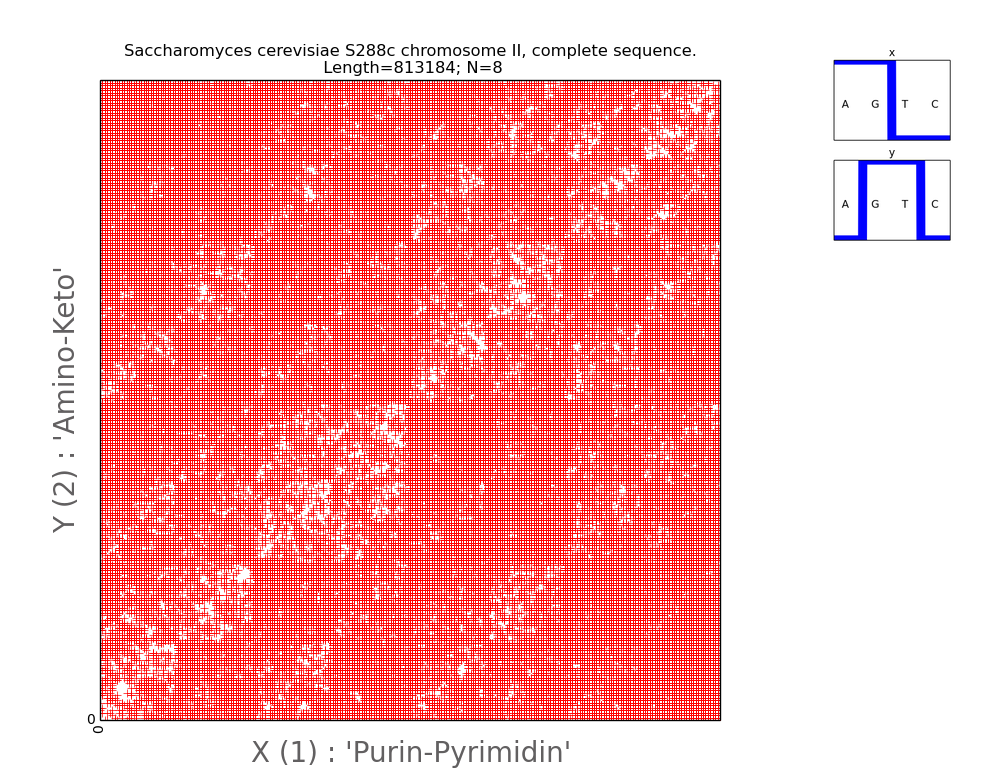
place in the fractal pattern. In Fig. 7, the visualization of the nucleotide
composition of the second chromosome of the single-celled microscopic fungus
"baker's yeast" shows a different behavior of the diagonal elements:
the diagonals are well traced only in one direction, the fractal repetitions of
the diagonals are also expressed only in one direction, and they display the
absent N-meters. The opposite diagonals responsible for the present N-meters
are less clearly traced.
Note
that diagonals and other elements of the pattern can be directed in different directions
in different organisms while maintaining the general structure of the pattern.
This feature can be simulated by reading the complementary DNA filament.
Fig.6.
Illustration
of two-dimensional representation of the nucleotide composition of the
bacterial genome. A pair of Walsh functions used for parameterization is
displayed in the upper right corner. The axes of abscissa and ordinate
correspond to the decimal representation of the binary coding of each 8-wire.
Fig.7.
Illustration
of two-dimensional representation of nucleotide composition of the second
chromosome of single celled microscopic fungus (baking yeast). A pair of Walsh
functions used for parameterization is shown in the upper right corner. The
axes of abscissa and ordinate correspond to the decimal representations of the
binary coding of each 8-wire.
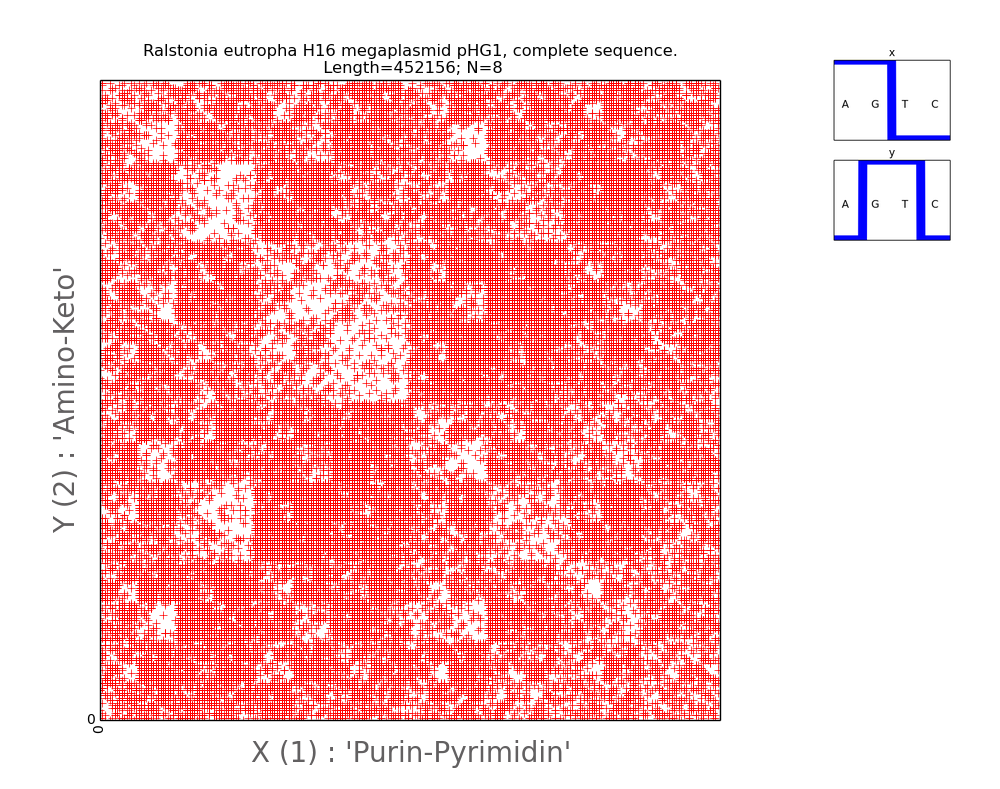
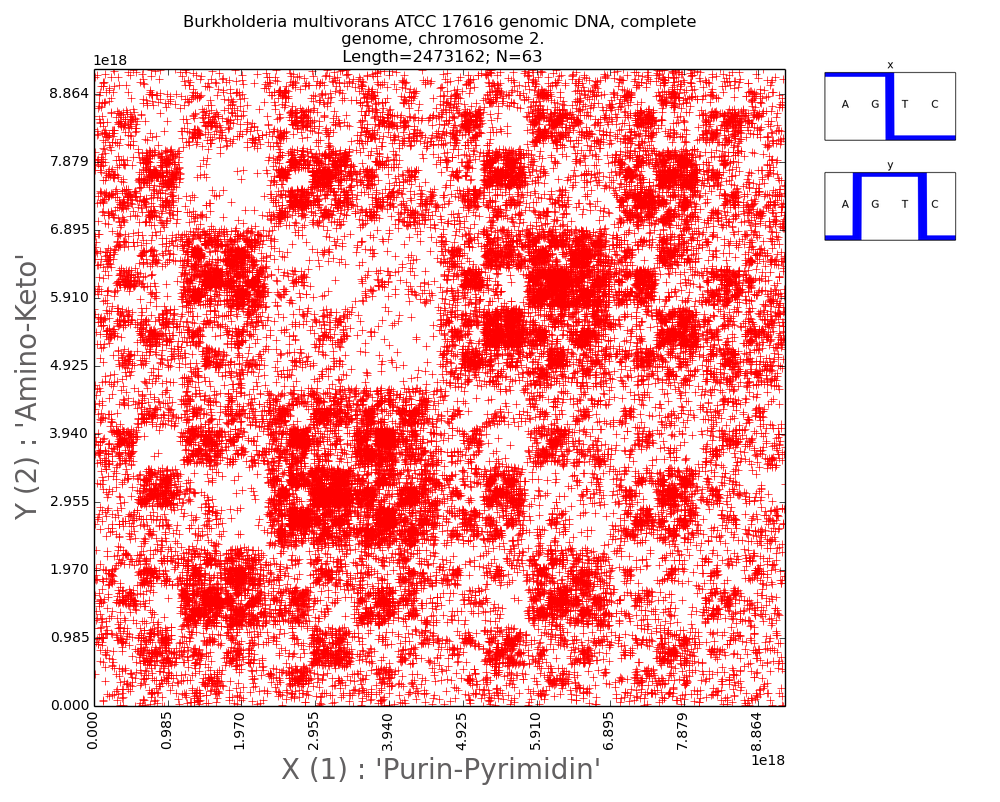
Figures 8 and 9 present
visual two-dimensional representations of Ralstonia eutropha (H16 megaplasmid
pHG1) and the complete genome of the Burkholderia multivorans protobacter, respectively.
Their visual patterns are characterized by pronounced fractality, and the
pattern of protobacteria has a bright form - the balance of present and absent
63-dimensions in its DNA is clearly visible (Fig. 9).
General scientific
methods of studying nucleic acids, as a rule, concentrate their attention on
those fragments that are present in them. The proposed method allows to present
in a clear form the phenomenology and features of the deficit and presence of
different types of N-mers. The absent and present N-dimensions of the
protobacterial genome in Fig. 9 make up a beautiful fractal. Thus, the geometric
approach allows to display the balance of present and absent 63-merials forming
structured fractal clusters in Figs. 8 and 9.
Fig.8.
Illustration of two-dimensional representation of the
nucleotide composition of the bacterial genome. The axes of abscissa and
ordinate correspond to the decimal representation of binary coding of each
8-wire. One of the characteristic patterns having fractal nature.
Fig.9.
Illustration of
two-dimensional representation of nucleotide composition of the second
chromosome of protobacteria. It can be seen that in this organism the present
and absent 63 wraps form a symmetrical fractal mosaic, the structure of which
is stable with respect to the reversal of flowers. To axes of abscissa and
ordinate there correspond decimal notions of binary coding of each 63-platform.
The conducted researches and analysis of
visualizations of nucleotide sequences of different kinds of living organisms
confirm that nucleotide composition can be identical in organisms which are not
related in the phylogenetic tree and different in related organisms [12]. A special
class of symmetries implemented in long DNA sequences of different organisms is
known. In work S.V. Petukhov [22] fractal genetic networks are resulted and
tetragroup symmetries are described. Thus, the known scientific data on fractality
of DNA are visually displayed on the basis of the offered method.
Fig. 10 shows an example of
integral-two-dimensional representation of human chromosome nucleotide composition
on one of the visualization planes. Examples of genetic mosaics built in
non-position number system are given in [9].
Fig.10.
Illustration of integral-double
representation of human chromosome nucleotide composition on one of the
visualization planes. A pair of Walsh functions used for parameterization is
shown on the right. The axes of abscissa and ordinate correspond to the number
of units of each 64 wafer using a pair of binary-opposition subalphabets.
Preliminary
results of the two-dimensional visualization method allow to draw a conclusion
about high stability of the final mosaics at noise of the initial sequence,
including at shifts of the sequence reading frame, in cases of removal of arbitrary
fragments of the sequence (thinning), at reversing of the whole analyzed chain
or its fragments, at different types of rearrangements of N-mers and
nucleotides (in some cases up to complete rearrangement of all nucleotides in
the sequence). In particular, the stability of mosaic patterns was observed at
removal of every second nucleotide, every third nucleotide, etc. In this case,
the visualization of nucleic acids in two-dimensional spaces in a number of
cases is characterized by pronounced symmetry and stability not only to noise
in the original data, but also to different values of the parameter N scale
within a certain range - this effect can be seen in the animations:
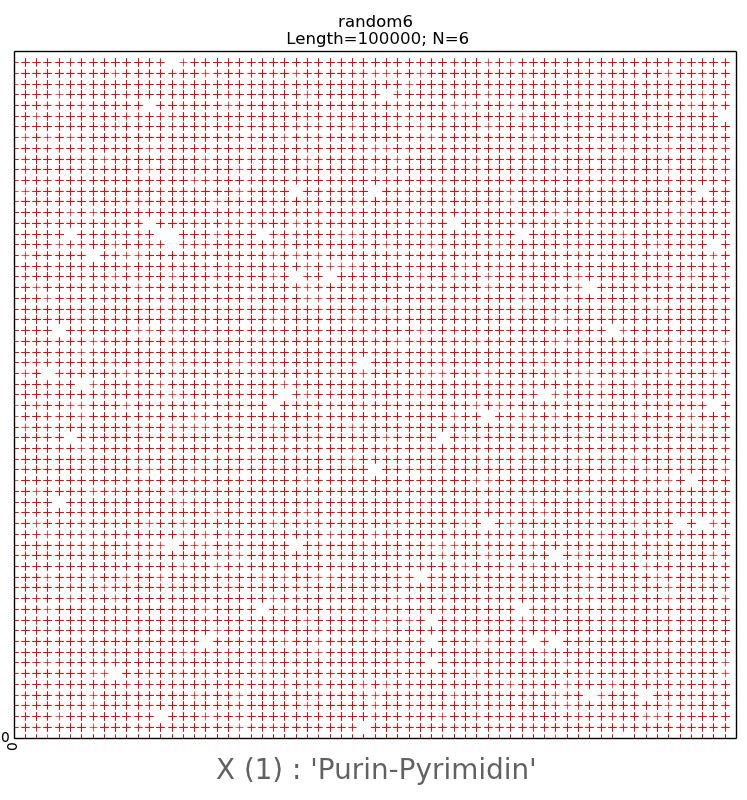
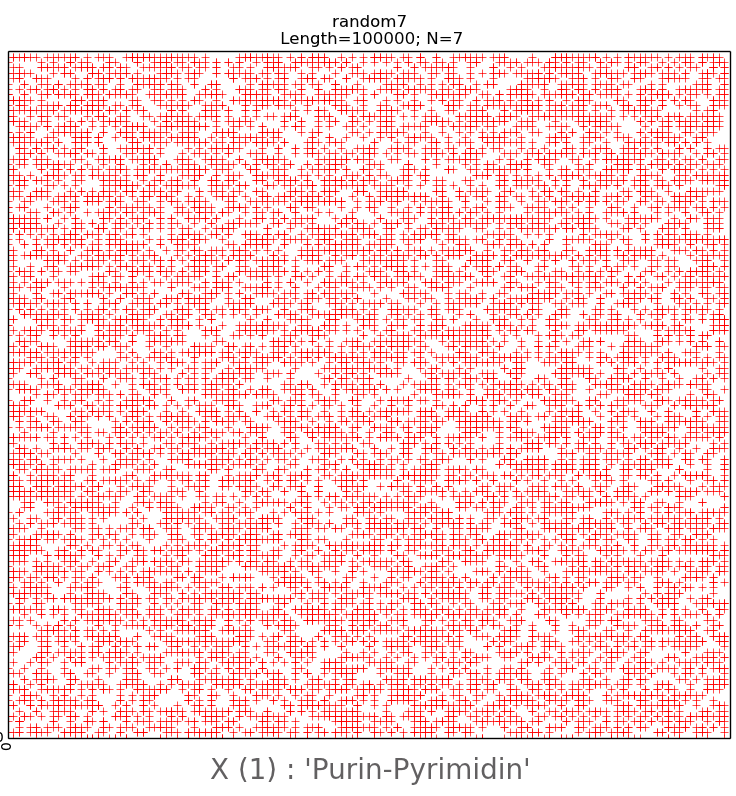
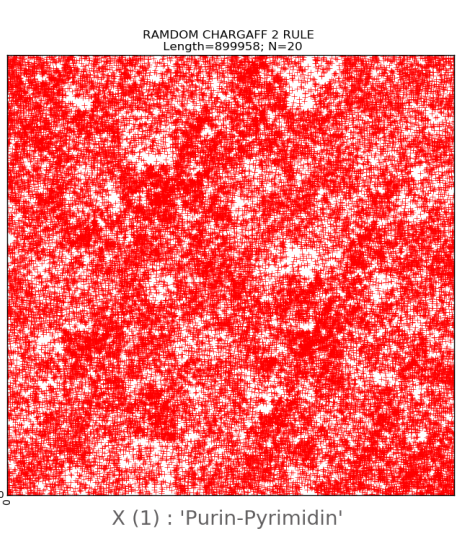
For further research, random sequences of nitrogenous
bases with a length of 100,000 nucleotides were created with the help of the
developed computer program, divided into N-brands of 8, 16 and 28. The randomly
generated sequences during visualization gave a pattern with all points
scattered chaotically (Fig. 11, upper row). Their visual representations are
irregular, chaotic in nature with the complete absence of any mosaics on all
subalphabets, which significantly distinguishes them from real long nucleotide
sequences.
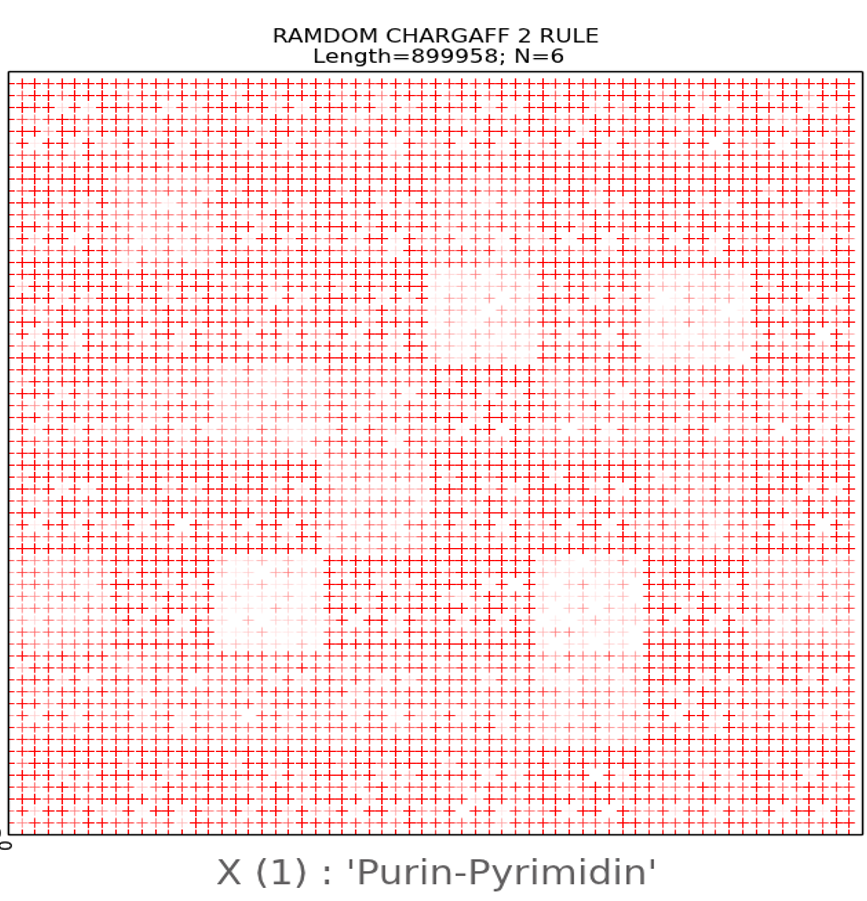
We have also created pseudo-random nucleotide sequences
on the computer, observing the second Chargaff rule, valid for each of the two
strands of DNA [1,11]. Fig. 11 shows a comparison of the sequences that were
randomly created without observing (bottom row) and with observing (top row)
the second Chargaff rule. For these sequences a special type of regularities at
different values of N, equal to 6.7 and 20 was visualized. From Fig. 11 we can
see that the random pattern, built by the second rule of Chargaff, is structured
due to the presence of empty flat areas, which are evenly distributed and
especially clearly visible at N = 6 in Fig. 11 in the lower row on the left. At
the same time, as noted above, a random pattern created without observing the
Chargaff ratio has a chaotic character in the visualization (upper row). From
this it is possible to draw a conclusion about geometrical connection of visualization
patterns by author's algorithm with algebraic rules of Chargaff.
Fig.11.
The upper
row is an illustration of a two-dimensional representation of the composition
of a randomly generated nucleotide sequence without following the Chargaff
rules. The lower row is an example of two-dimensional representation of the
nucleotide composition of a randomly generated sequence taking into account the
second Chargaff rule. The abscissa and ordinate axes correspond to decimal
representations of the binary coding of each N-platform.
Thus, two-dimensional
visualization of chains of nitrous bases allows to display variants of
performance of quantitative rules of Chargaff [1,11] with application of the
apparatus of final geometry [21]. This fact can help in the study of internal
symmetries and other characteristics of nucleic acids to study complex
relationships between living organisms.
We have constructed
visual representations of DNA of different kinds of penicillin. The obtained
results testify that genomes of this group, as a rule, generate mosaics of high
density resembling mosaics of random sequences, which testifies to the high
diversity of nucleotide composition. Perhaps the medical value of penicillin is
related to this particular feature.
Thus, two-dimensional
imaging methods seem useful for studying hidden patterns in chromosomes, as
well as for classification and comparative analysis of different genomes with
possible applications in biotechnology and medicine.
As
noted, binary subalphabets are linked by an addition operation on the module
two and set the space with properties in which the coordinates of each point
are linked. In this regard, it makes sense to consider each dimension separately.
There are three parametrically one-dimensional linked visualisation spaces.
Using parametrically one-dimensional coordinate axes {X}, {Y} and {Z} gives
three different mappings using corresponding sub-alphabets. The abscissa axis
encodes the serial number of the N-platform, the ordinate axis encodes the
ascending ordered decimal values of the binary representation of each
N-platform (note: the visualization itself is two-dimensional, but the
parametric measurement is one).
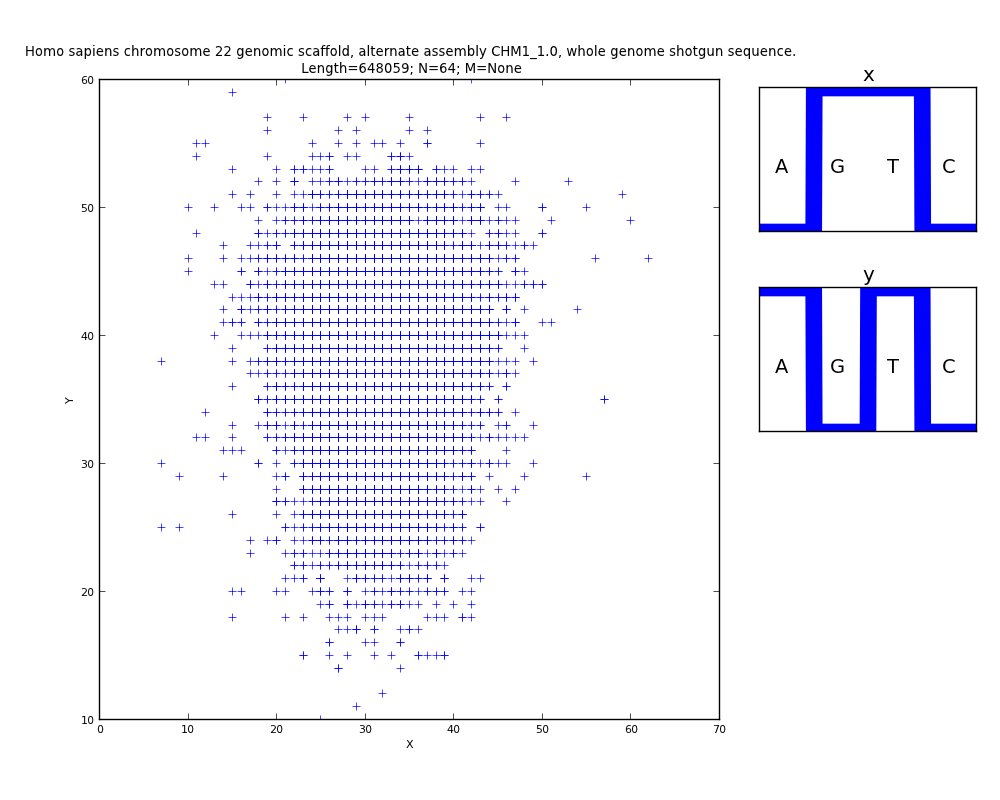
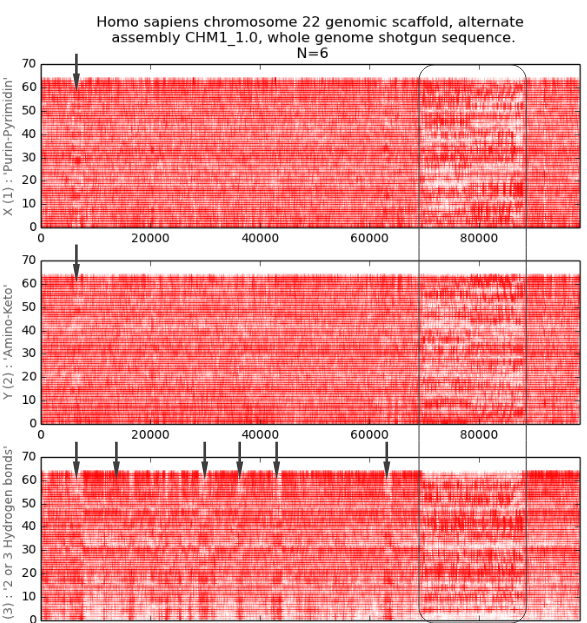
Figure
12 shows an example of visualization of a human chromosome where areas with
different nucleotide composition are clearly visible. These specific regions
are marked with arrows in the figure and can be visualized at different scales
in two-dimensional imaging spaces for their detailed analysis.
Fig.12.
Visualization
of the three-channel representation of the nucleotide composition of the 22nd
chromosome fragment of Homo Sapiens. Each of the three projections corresponds
to a binary-opposition sub-alphabet. In each channel the abscissa axis encodes
the ordinate number of the N-platform, the ordinate axis encodes the ascending
ordered decimal values of the binary representation of the N-platform. The
arrows highlight some areas with different nucleotide composition. A large area
with different nucleotide composition is circled. We can see that in different
parts of the chromosome the nucleotide composition may differ for each of the
channels.
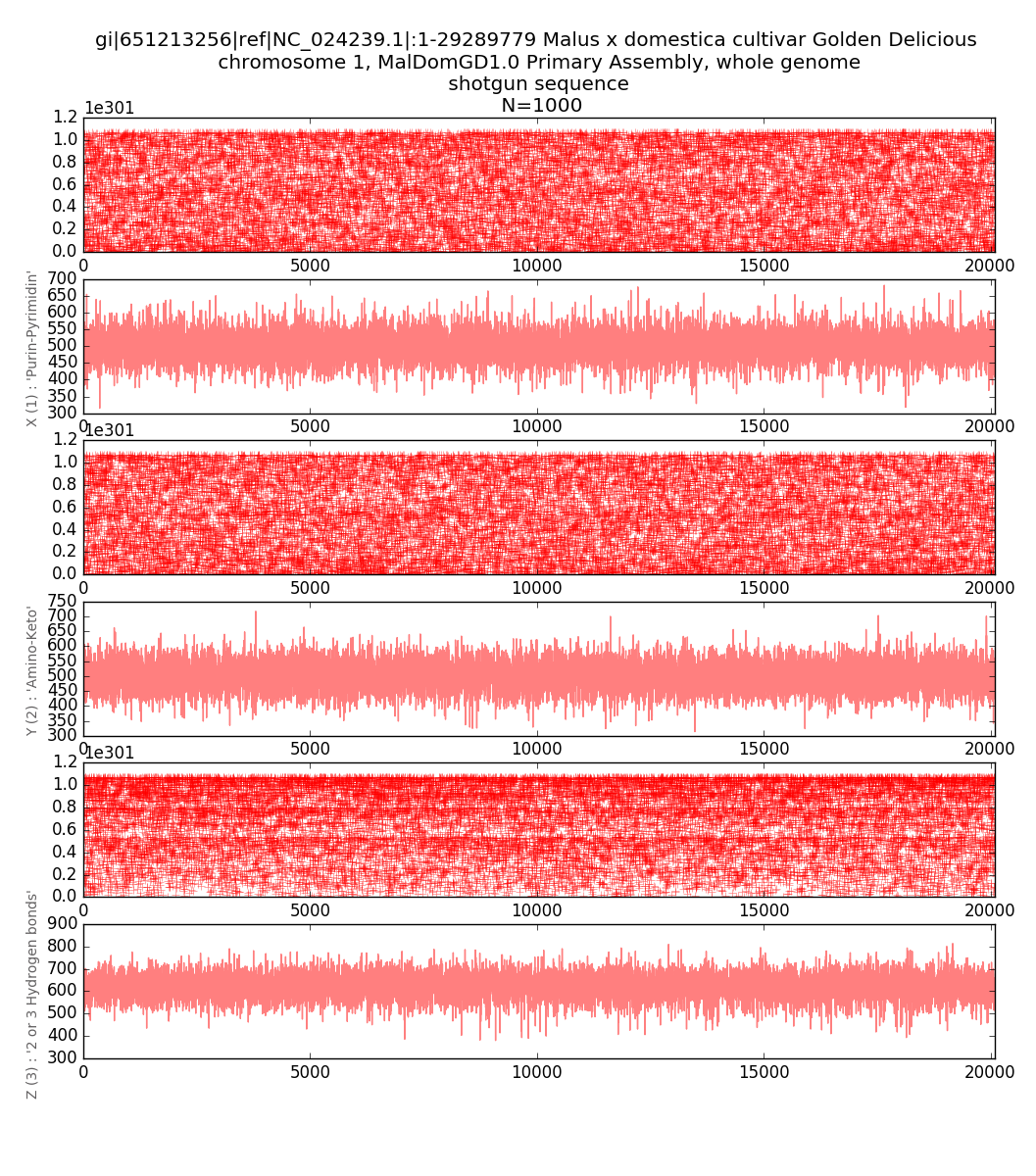
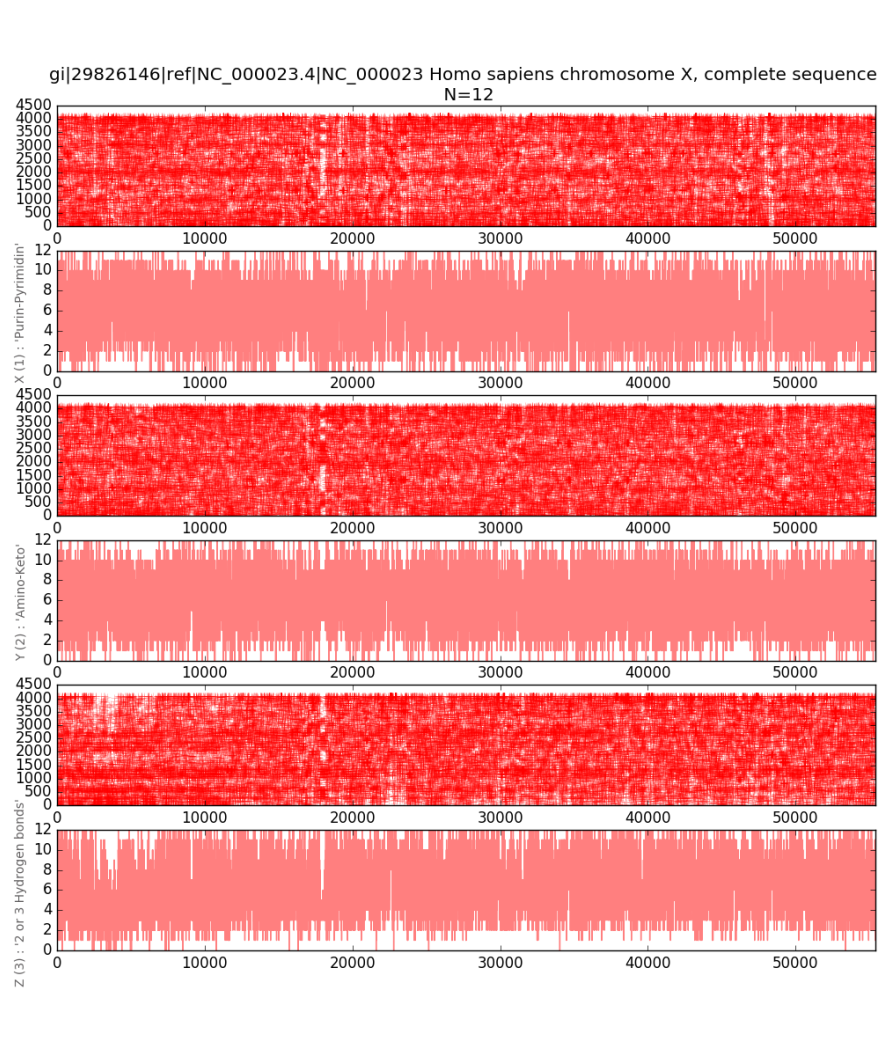
In Figures 13 and 14, an integral one-dimensional
visualization of the total number of units in N-dimensional codes is additionally
given for each of the three sub-alphabets. The resulting graphs allow to
estimate changes in the nucleotide composition when reading a fragment of a
molecule from beginning to end. The depth of registered changes is determined
by the scaling parameter N.
Fig.13.
Visualization
of the three-channel representation of the nucleotide composition of the apple's
1st chromosome fragment. Each of the three rows corresponds to a
binary-opposition sub-alphabet. The abscissa axis encodes the serial number of
1,000-plet, the ordinate axis encodes the number of units in 1,000-plet.
Fig.14.
Visualization
of the three-channel representation of the nucleotide composition of the human
X chromosome. Each of the three rows corresponds to a binary-opposition
sub-alphabet. The abscissa axis encodes the serial number of the 12-platform,
the ordinate axis encodes the number of units in the 12-platform.
Parametric
one-dimensional imaging methods are convenient because they allow to display
the nucleotide composition of the chromosome, as it is impossible to display it
in two-dimensional and three-dimensional projections. In this regard, the
described one-dimensional imaging methods seem to be informative and promising
for further studies.
It
should be noted that three-channel representation is combined with the
classical theory of color perception (RGB), in which it is considered that the
eye perceives three basic colors: red, green and blue, and combinations of
the three basic colors can get the rest of the colors. This theory is mentioned
in [15] in connection with genetic algebra. Each of three channels of
one-dimensional visualization can be compared to one of three basic colours.
Intensity of colour of each point of two-dimensional visualisation is various, therefore
two-dimensional and three-dimensional representations allow to consider combinations
of colours. It allows to strengthen color perception in genetics and opens new
possibilities for parametric visualization according to the stated method
(however, our experiments showed that it considerably increases counting time).
For
the author's method of parametric imaging it is proposed to introduce a new
term "genetic geometry" or "genometry" as the basis for the
corresponding scientific direction in the field of molecular-biological
biosemiotics [19].
The
result of the study is the achievement of the goal to develop methods of visualization
of long nucleotide sequences. The connections of molecular genetic systems with
Hadamard's binary number system and matrixes are demonstrated. The hypothesis
about the possibility of visualizing the internal symmetries in the nucleotide
composition was confirmed. Nucleic acids have a visual representation.
Parametric visualization of both fragments and entire molecules of DNA and RNA
allowed to substantiate their connection with geometric mosaics of different
types (see, for example, Fig. 4-9). The proposed method allows to estimate the
types of relations between present and absent N-meters in DNA of different organisms
(these relations can be characterized by fractal-cluster organization, a vivid
example - Fig. 9). The scaling parameter N makes it possible to investigate the
genome at many levels of detail to find hidden symmetries and regularities.
The
emergence of reasonable methods for comparing geometric representations of
genotypes with certain phenotypic features expands the methods of research in molecular
genetics. In addition, it opens up the possibility of modeling pseudo-random
nucleotide sequences with observance of the phenomenological rules of Charghaff
for their visualization and further research. Large-scale parametric visualization
of the nucleotide composition contributes to a deeper understanding of genetic
phenomena not only by simplifying perception, but also by using adaptive neural
network technologies, as the structure of chromosomes of living organisms,
represented in the binary code, corresponds to the format of binary artificial
neural networks [13].
The
author's method of visualization is an additional criterion of classification
and detection of interspecific relationships. In this regard, modern ontologies
and thesauruses for organization and storage of molecular genetic data can be
equipped with visualization options for educational purposes, as well as for
presentation and search of biological information. The proposed method can also
help advance the understanding of the principles of the immune system in
recognizing the nucleotide composition of viruses, DNA of parasites, as well as
in food chains and ecosystems. Geometric concepts can help in the study of
point mutation mechanisms and CRISPR-Cas systems [16]. It becomes possible due
to visual interpretation of basic characteristics of polynucleotide fragments
of a certain nucleotide composition with visualization of the final geometry
and structure of the genetic code.
The
presented results allow us to speak about the author's methods of nucleic acids
visualization as a scale-parametric model of DNA, which complements the structural
model of the double helix of J. Watson and F. Creek [17,18].
The
author expresses his gratitude to Sergey Valentinovich Petukhov, Vitaly Ivanovich
Svirin, Konstantin Vladimirovich Pleshakov, Denis Sergeevich Izyumov and Dmitry
Vitalievich Salonin for fruitful scientific discussions.
1. Chargaff E, Lipshitz R, Green C (1952). "Composition of the deoxypentose nucleic acids of four genera of sea-urchin" (PDF). J Biol Chem.195 (1): 155–160. PMID 1493836
2. S.V.Petoukhov, M.He. Symmetrical Analysis Techniques for Genetic Systems and Bioinformatics: Advanced Patterns and Applications. 2010, Hershey, USA: IGI Global. 271 p.
3. N.A.Balonin, Y.N.Balonin, D.Z. Djokovic, D.A. Karbovskiy, M.B.Sergeev. Construction of symmetric Hadamard matrices https://arxiv.org/abs/1708.05098
4. Georgiou, S.; Koukouvinos, C.; Seberry, J. (2003). "Hadamard matrices, orthogonal designs and construction algorithms". Designs 2002: Further computational and constructive design theory. Boston: Kluwer. pp. 133–205. ISBN 1-4020-7599-5.
5. I.V. Stepanian, S.V. Petoukhov. The matrix method of representation, analysis and classification of long genetic sequences http://arxiv.org/pdf/1310.8469.pdf
6. H., Harmuth Applying of methods of theory of information in phisics / - Moscow.: Mir, 2016. - p. 344.
7. Ferleger, Sergei V. (March 1998). RUC-Systems In Non-Commutative Symmetric Spaces (Technical report). MP-ARC-98-188.
8. Jeffrey H.J. (1990). Chaos game representation of gene structure. - Nucleic Acids Research, Vol.18, No.8, p. 2163-2170.
9. Feldman, David P. (2012), "17.4 The chaos game", Chaos and Fractals: An Elementary Introduction, Oxford University Press, pp. 178–180, ISBN 9780199566440.
10. G. Darvas, A.A. Koblyakov, S.V.Petoukhov, I.V.Stepanyan. Symmetries in molecular-genetic systems and musical harmony // Symmetry: Culture and Science Vol. 23, No. 3-4, 343-375, 2012 http://symmetry.hu/scs_online/SCS_23_3-4.pdf
11. Rudner, R; Karkas, JD; Chargaff, E (1968). "Separation of B. SubtilisDNA into complementary strands. 3. Direct analysis". Proceedings of the National Academy of Sciences of the United States of America. 60(3): 921–2.doi:10.1073/pnas.60.3.921. PMC 225140. PMID 4970114.
12. Townsend JP, Su Z, Tekle Y (2012). "Phylogenetic Signal and Noise: Predicting the Power of a Data Set to Resolve Phylogeny". Genetics. 61(5): 835–849.doi:10.1093/sysbio/sys036. PMID 22389443.
13. Stepanyan I.V., Ziep N.N. Growing convolutional neural-like structures for problems of recognition of static images // Neurocomputers: development, application. 2018. №5. pp. 4-11.
19. Sharov A. (1992). Biosemiotics: functional-evolutionary approach to the analysis of the sense of information. In: Biosemiotics: The Semiotic Web 1991. T.A.Sebeok and J.Umiker-Sebeok (eds.), 345-373. Berlin: Mouton de Gruyter.
20. Waterman M.S. Introduction to Computational Biology. Map, Sequences and Genomes. London: Chapman & Hall, 1995. xvi + 432 pp.
21. Batten, Lynn Margaret (1997), Combinatorics of Finite Geometries, Cambridge University Press, ISBN 0521590140
22. Petoukhov S.V., Petukhova E.S., Svirin V.I. New Symmetries and Fractal-Like Structures in the Genetic Coding System. – Advances in Intelligent Systems and Computing, v. 754, 2018, p. 588-600, https://doi.org/10.1007/978-3-319-91008-6_60
23. Mcdonnell K, Waters N, Howley E, Abram F. Chordomics: a visualisation tool for linking function to phylogeny in microbiomes. Bioinformatics. 2019;
24. Mathema VB, Dondorp AM, Imwong M. OSTRFPD: Multifunctional Tool for Genome-Wide Short Tandem Repeat Analysis for DNA, Transcripts, and Amino Acid Sequences with Integrated Primer Designer. Evol Bioinform Online. 2019;15:1176934319843130.
25. Iacoangeli A, Al khleifat A, Sproviero W, et al. DNAscan: personal computer compatible NGS analysis, annotation and visualisation. BMC Bioinformatics. 2019;20(1):213.
26. Martens KJA, Van beljouw SPB, Van der els S, et al. Visualisation of dCas9 target search in vivo using an open-microscopy framework. Nat Commun. 2019;10(1):3552.
RUSCOMNADZOR Reg. Number El. № ФС77-37344 INFORMREGISTR Reg. Number № 0421100125
Copyright http://sv-journal.org