Citation: R. Mavrevski, M. Traykov. Visualization software for Hydrophobic-polar protein folding model (2019). Scientific Visualization 11.1: 11 - 19, DOI: 10.26583/sv.11.1.02
The simplest and most used models of protein folding is the Hydrophobic-Polar (HP) model. The HP model labeling the amino acids as Hydrophilic (H) or Polar/ydrophilic (P). The folding of amino acids sequence is configured as self-avoiding walks on the 2D or 3D lattice, where the optimal conformation has maximum number of contacts between H amino acids (H-H contacts) that are not adjustment in amino acid sequence. In this paper, we develop and present software for visualization of HP protein folding problem under the HP model on the 2D square lattice. For the development of HP folding visualization software, we used MS Visual Studio, .NET Framework 2.0 and C# language. If we have HP sequence and folding results for this sequence, obtained by optimization software as CPLEX or GUROBI, then using the 2D visualization software we can visualize the obtained results in square lattice. This visualization software is a valuable tool for the study of HP folding and is a great pedagogic instrument. All figures of HP folding included in this paper are actual screenshots of our visualization program.
Keywords: protein folding, HP model, visualization.
The proteins
are major and important class of biological macromolecules, which perform
structural and catalytic functions in the muscle, skin, hair, etc. The protein
is a chain where each element is one of 20 different amino acids [1-3]. An
important property of proteins is their hydrophobicity, which is expressed in
how much they are repel by the water. Understanding the processes leading to a
protein folding into its functional state is the important problem in molecular
biophysics. This determines how the protein chain folds into a complex
conformation (conformation of the molecule). This conformation is stable, has small
free energy and is very important for the function of the protein. It is
important to note that the amino acid sequence is the primary structure. In the
literature there are described different types of algorithms to predict the
tertiary structure of proteins [4]. All these algorithms use an abstract model
of real proteins, describing their characteristics. Most common models are the
lattice models, which use a 2D or 3D lattices to describe the positions of the
amino acids and an energy function that needs to be minimized according to positions
of the amino acids in the lattice. This is the so-called Protein folding
problem. The key model to solve the Protein folding problem is Dill's Hydrophobic-Polar
(HP) model (so-called HP folding problem) that tries to form hydrophobic core,
i.e. tries to put hydrophobic amino acid in the center of obtained fold
surrounded by polar amino acid [5-9].
In the literature,
there are many mathematical models described and algorithms for Protein folding
problem or HP folding problem. However, the most articles describing
mathematical models and algorithms for HP folding lack mentioning the
visualization of the obtained results (folds). In this paper, we propose
software for visualization of received folds in HP folding problem under 2D
square lattice after we solve the problem using some optimization software, as
a CPLEX or GUROBI. In the section 3 (Results) we present our experiments and results.
Thus, it is possible to make detail analysis of the folding process, e.g. by comparison
of different conformations for one input HP sequence or identifying the
connectivity between conformations. Visualizing the folding paths (so-called
self-avoiding paths), we can study different regions of the protein
conformation, as well as determine the difficulty in folding of different
proteins. Visualizing the folding is significant challenge owing to the
similarity to nature state of the proteins. The other motivation to investigate
and visualize the protein folding process in details is the potential help in
understanding the role of protein functions [7, 8].
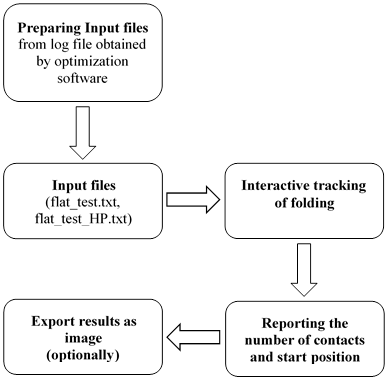
To solve the Protein
folding problem in 2D or 3D HP model we generate .lp file (https://www.ibm.com/support/knowledgecenter/SSSA5P_12.5.0/ilog.odms.cplex.help/CPLEX/FileFormats/topics/LP.html)
that contains description of a mathematical model for the problem. After that, we
submit the obtained .lp file to MIP (Mixed Integer Programming) solver like
CPLEX (https://www.ibm.com/analytics/cplex-optimizer)
or GUROBI (http://www.gurobi.com/)
and run the solver. When the solver finishes, we obtain a log file that
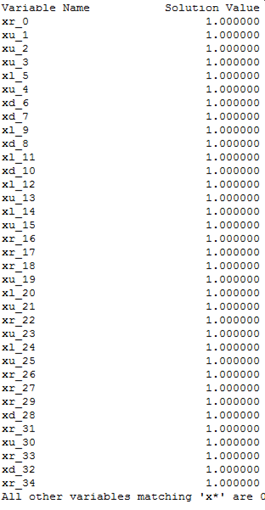
contains solution to the mathematical model (see Figure 1). The obtained log
file can be stored in different formats (according to the model description in
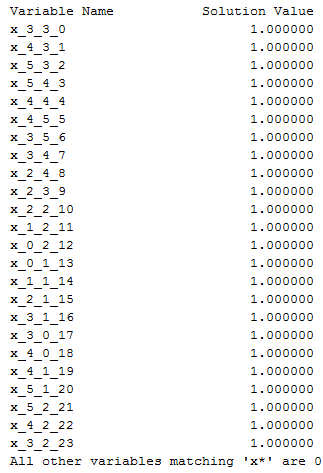
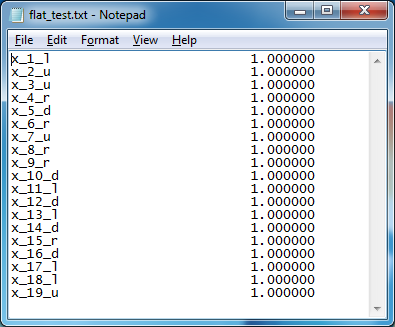
.lp file), i.e. we do not know the format of the file so wetransform the log file manually (using Notepad++
software) to the appropriate input file (Figure 2) with the “.txt” extension
(named “flat_test.txt”) for our visualization software. In the future, we plan
to automate the transformation process.
The first
column of the transformed file contains the variable names and the
corresponding direction of movement (r – right, l – left, u – upper, d – down)
during the folding process. The second column contains solution value for the
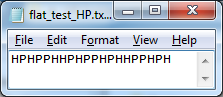
corresponding variable. In addition, we prepare manually (using Notepad++ software)
second input file for our HP Folding Visualization software (named
“flat_test_HP.txt”) containing the HP sequence (see Figure 3).
In this article,
we show software called “HP Folding Visualization” developed by us (see Figure
4 and 5). The aim of this software is to make visual representation of
performers of HP protein folding in 2D lattice. We can run our software as
desktop application, i.e. we do not need to make installation, just copy and
paste the folder with software files on our computer. To develop the “HP Folding
Visualization” software we use MS Visual Studio, .NET Framework 2.0 and C#
language. To create a simple and flexible grid to use in all cases where it is
necessary to visualize HP folding process we used additional library “SourceGrid.dll”
(https://archive.codeplex.com/?p=sourcegrid).
There are available many controls of this type, but they are expensive,
difficult to customize or not compatible with .NET. SourceGrid is free Windows
Forms control written entirely in C#.
VS.NET is one
of the world's leading Integrated Development Environment (IDE). With its help,
we can do each of the typical tasks related to building an application –
writing code, creating user interface, compiling, running and testing,
debugging, error tracking, viewing documentation and others [10, 11]. The
Microsoft .NET Framework is platform created by Microsoft that provides a
programming model, library of classes, Framework Class Library (FCL), and
Common Language Runtime (CRL). .NET applications are written in high-level
languages (C#, VB.NET, C ++ / CLI, etc.) and compiled into a
platform-independent intermediate language called Common Intermediate Language
(CIL). In this work, we used the C#
language. During execution, the CIL code is automatically compiled by CLR for
the specific hardware platform and current operating system.
Fig.
4. Conceptual scheme of “HP Folding Visualization”
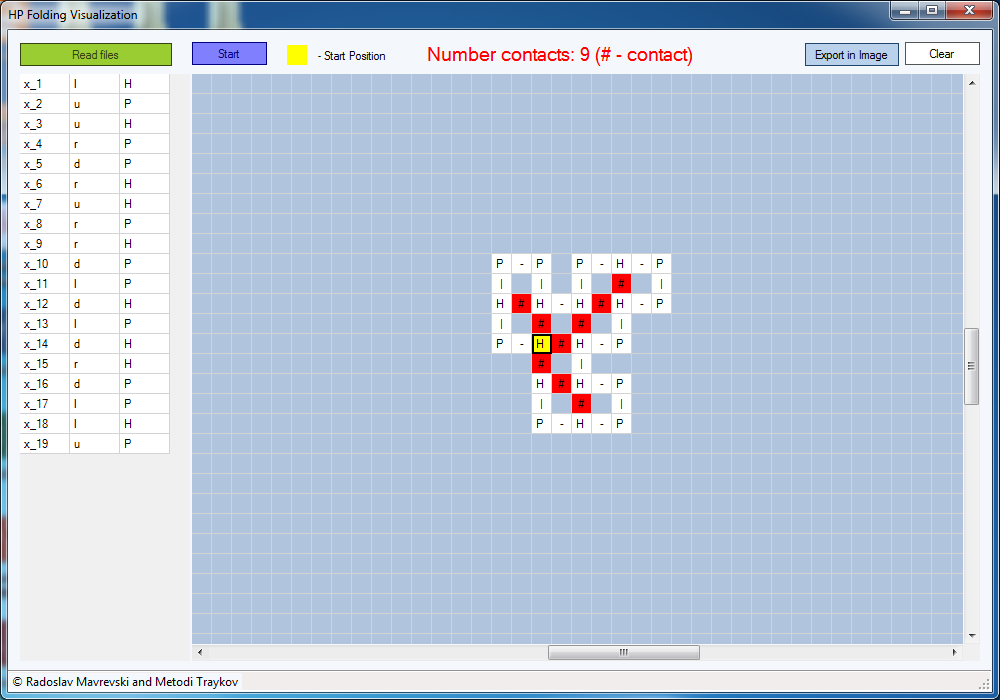
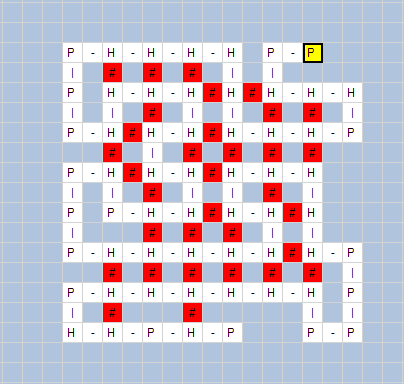
Using our software,
we can interactively track the folding process (Figure 5), i.e. each movement
of the amino acids in the 2D square lattice, H-H contacts are marked as “#” and
colored in red.
Fig. 5. Interactively
track the folding process
The start position is
marked with yellow color. With dash we indicate the adjacent amino acids in the
input HP sequence (see Figure 6).
Fig.
6. The main window of “HP Folding Visualization” software
In this article,
we described the software “HP Folding Visualization” for 2D visualization of
the HP folding developed by us. For our examples, we use output files from
CPLEX that are solutions of mathematical models described in .lp files. These
output files contain obtained folds for HP sequences with different length [9].
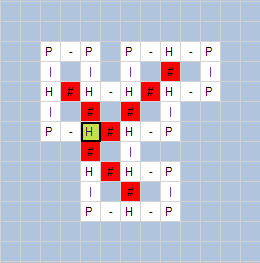
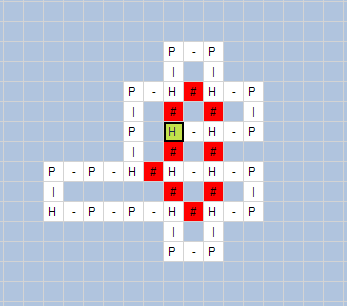
The next
figures show obtained results by using “HP Folding Visualization” software.. In
Figure 7 we show obtained folds for HP sequences with length:
(a) 20 amino
acids – HPHPPHHPHPPHPHHPPHPH;
(b) 24 amino
acids – HHPPHPPHPPHPPHPPHPPHPPHH;
(c) 60 amino
acids – P P H H H P H H H H H H H H P P P H H H H H H H H H H P H P P P H H H H H H H H H H H H P P
P P H H H H H H P H H P H P.
(a)
(b)
(c)
Fig. 7. Visualization of HP folding by “HP Folding
Visualization” software: (a) protein with length 20, (b) protein with length
24, (c) protein with length 60.
In the above
figures, we can see the H-H contacts (the contact between H amino acids that
are not adjacent in the protein sequence) and their number.
All data for
our input files are obtained using algorithm described by Yanev et. al [9].
This algorithm uses mathematical model (Integer Programming) for HP model in 2D
square lattice and as we said above, we solve this model with MIP solver CPLEX.
The CPLEX solution file (.sol or .log) contains solution variables (binary
type) (see Figure 1) for the objective function described in .lp file, i.e. the
input for “HP Folding Visualization” software is file that contains binary
variables (number of binary variables is equal to number of amino acids). This
is the reason why we cannot compare our software with other bioinformatics
visualization tools (e.g. Bioblender (http://www.bioblender.org/),
PEP-FOLD (http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/),
PyMOL (https://pymol.org/),
ProtSkin (http://www.mcgnmr.mcgill.ca/ProtSkin/)
etc.) because they use input files as .pdb, .cif, .xml, etc. To run the input file
for “HP Folding Visualization” software in mentioned above bioinformatics
visualization tools we need to convert the CPLEX solution file (with binary
variables) to .pdb, .cif or xml file that contains specific information (as
distances in Ångström etc.) for protein.
On the other hand,
the bioinformatics visualization tools, like Bioblender and PEP-FOLD, cannot visualize the
results obtained by algorithms that use Integer Programming approach (like
Yanev et al. algorithm) [9, 12]. To work correctly most bioinformatics
visualization tools need .pdb or FASTA files, protein PDB ID, specific
coordination or original amino acid sequence (not HP sequence), i.e. they
cannot visualize folds obtained by Integer Programming approach (2D or 3D
lattices HP model) and algorithms as mentioned above.
The
computational experiments were made in simple 2D lattice HP model. This model
is used by Yanev et al. to solve HP folding problem, so discussed folds are
proven and described in [9]. In particular, our software may be helpful to
probe details of folding trajectories and number of contacts in the study of
proteins folding. Using our software, we expect to be able to shed light into
the nature of these various conformational states. In addition this article can
help the students in secondary and higher schools to acquire additional skills
in bioinformatics and to learn more about the proteins and their folding.
Ahead of us
stands the challenge to use our software together with other methods that
efficiently visualize the HP folding in different lattice types.
If you want to
use "HP Folding Visualization" software please send us mail on radoslav_sm@abv.bg.It will be pleasure for us to provide
our software.
1. Gō N,
Taketomi H: Respective roles of short- and long-range interactions in protein
folding. Proceedings of the National Academy of Sciences of the United States
of America 1978, 75:559-563.
2. Dill KA:
Theory for the folding and stability of globular proteins. Biochemistry 1985,
24:1501-1509.
3. Lau KF,
Dill KA: A lattice statistical mechanics model of the conformational and
sequence spaces of proteins. Macromolecules 1989, 22:3986-3997.
4. Istrail,
S., and Lam, F. 2009. Combinatorial algorithms for protein folding in lattice
models: A survey of mathematical results. Commun. Inf. Syst. 9, 303–346.
5. Dill KA,
Bromberg S, Yue K, Fiebig KM, Yee DP, Thomas PD, Chan HS: Principles of protein
folding - A perspective from simple exact models. Protein Science 1995,
4:561-602.
6. Thalheim T.,
Merkle D., Middendorf M. Protein Folding in the HP-Model Solved With a Hybrid
Population Based ACO Algorithm. IAENG International Journal of Computer Science
2008, 35:391-300.
7. Shan Y,
Arkhipov A, Kim ET, Pan AC, Shaw DE (2013) Transitions to catalytically
inactive conformations in EGFR kinase. Proceedings of the National Academy of
Sciences of the United States of America 110: 7270–7275.
8. Reddy AS,
Wang L, Singh S, Ling YL, Buchanan L, et al. (2010) Stable and metastable
states of human amylin in solution. Biophysical Journal 99: 2208–2216.
9. Yanev N.,
Traykov M., Milanov P., Yurukov B. Protein Folding Prediction in a Cubic
Lattice in Hydrophobic-Polar Model, Journal of Computational Biology, 2017,
24(5), 412-421
10. Arora, G.,
Aiaswamy, B., Pandey, N. Microsoft C# Professional Projects, Portland, United
States, Premier Press, 2002.
11. Krishna,
P. "Announcing the .NET Framework 4.7.1". .NET Blog. Retrieved 17
October 2017.
12. Yoon, H.,
2006. Optimization Approaches to Protein Folding. Georgia: School of Industrial
and System Engineering, Institute of Technology
RUSCOMNADZOR Reg. Number El. № ФС77-37344 INFORMREGISTR Reg. Number № 0421100125
Copyright http://sv-journal.org