CLUSTVIS1: НОВЫЙ ПРОГРАММНЫЙ ПАКЕТ ДЛЯ ВИЗУАЛИЗАЦИИ АТОМНЫХ КЛАСТЕРОВ И МОЛЕКУЛ
А.Ю. Пищулина1, С.А. Шостаченко, М.М. Маслов, К.П. Катин, В.С. Прудковский2,3, Р.В. Рыжук, Н.И. Каргин
Национальный Исследовательский Ядерный Университет «МИФИ», Москва, 115409 Россия
1 Всероссийский научно-исследовательский институт по эксплуатации атомных электростанций, Москва, 109507 Россия
2 CNRS, LAAS, 7 avenue du colonel Roche, F-31400 Toulouse, France
3 University de Toulouse, LAAS, F-31400 Toulouse, France
Содержание
2. Визуализация трехмерной структуры кластера
3. Описание программы визуализации
4. Краткий обзор пользовательского интерфейса
5. Интеграция визуализатора со сторонними программами
Аннотация
В работе представлен новый программный пакет, предназначенный для статической и динамической визуализации структуры атомных кластеров и молекул. В отличие от аналогичных программных продуктов, представленный пакет скомпилирован в виде динамически подключаемой библиотеки. Это позволяет легко интегрировать его с любой сторонней расчетной программой для быстрой визуализации результатов расчета. Проведенная апробация подтверждает пригодность нового пакета для визуализации широкого класса атомных кластеров и наноструктур, включая биологические макромолекулы.
Ключевые слова: моделирование наноструктур, молекулярная динамика, структурная релаксация, атомная визуализация.
1. Введение
Широко используемые программные пакеты с открытым исходным кодом, предназначенные для атомистического моделирования (например, GAMESS [1], YAeHMOP [2], и др.), обычно не позволяют визуализировать структуру кластеров и молекул. Кроме того, исследователи часто разрабатывают свои собственные программы для реализации специфических алгоритмов, которые также не обеспечивают возможности визуализации результатов. Это приводит к необходимости использовать дополнительное программное обеспечение для наглядного представления выходной информации, в частности, структур атомных кластеров или молекул. Есть целый ряд программных продуктов, обеспечивающих трехмерную атомную визуализацию наноструктур. Примерами таких продуктов являются широко распространенные программы Chemcraft [3], Avogadro [4], Spartan [5] и др. Они обеспечивают множество возможностей для моделирования молекул и могут взаимодействовать с любой сторонней программой посредством файлового интерфейса. Однако такой интерфейс требует частого обращения к жесткому диску, что снижает производительность программного комплекса.
В этой статье мы предлагаем новую программу, предназначенную для визуализации структуры атомных кластеров и скомпилированную в виде динамической подключаемой библиотеки. Это позволяет ей легко интегрироваться с любыми сторонними программами, реализующими квантово-химические вычисления, независимо от языков программирования, на которых они написаны. Взаимодействие между сторонней программой и модулем визуализации может быть организовано не только посредством файлового интерфейса, но и исключительно через оперативную память. Это увеличивает скорость обмена данными и обеспечивает совместное использование памяти. Чтобы проиллюстрировать возможности нового визуализатора, мы объединили его с оригинальным алгоритмом сильной связи [6], предназначенным для моделирования полинитрокубанов.
2. Визуализация трехмерной структуры кластера
Основная задача программы визуализации – обеспечить возможность «вращения» атомного кластера, то есть просмотра его структуры с разных направлений. Кроме того, необходимы специальные функции для композиции нескольких вращений, так как может понадобиться изменить ориентацию кластера несколько раз. Для реализации вращения трехмерных структур существуют три общепринятых метода. Они основаны на углах Эйлера, матрицах вращения и кватернионах [7], соответственно.
Метод, основанный на углах Эйлера, имеет два недостатка. Во-первых, в рамках этого метода невозможно выполнить поворот вокруг некоторой оси независимо от предыдущих вращений вокруг двух других осей [8]. Во-вторых, метод неудобен для описания нескольких последовательных вращений. В этом случае приходится представлять вращения в другой форме, например, в матричной. Затем матрицы поворотов перемножаются для получения результирующей матрицы, которая затем преобразуется обратно в углы Эйлера [8-9].
Метод, использующий матрицы вращения, также имеет недостатки. Он обладает меньшей численной устойчивостью по сравнению с подходом, основанным на кватернионах. При матричном описании множества последовательных поворотов возможно накопление ошибок, связанных с ограниченной точностью компьютерного представления чисел [10].
По этим причинам для описания вращений кластера мы выбрали метод, основанный на кватернионах. К тому же он хорошо сочетается с функциями открытой графической библиотеки (OpenGL), поскольку в ней используются оси и углы поворота в качестве входных и выходных данных. Поскольку наша программа управляется при помощи компьютерной мыши, эти данные вычислялись на основе ее экранных координат.
3. Описание программы визуализации
Для реализации программы визуализации был выбран язык программирования C#, разработанный Microsoft для .NET Framework [11]. Также использовалась OpenGL спецификация, которая является подходящей библиотекой для рендеринга [12].
При визуализации кластеров используется представление «атом-связь» (атомы отображаются в виде сфер, а связи между ними – в виде соединяющих сферы цилиндров). Связь отображается только в том случае, если расстояние между двумя атомами не превышает 1.8 Å, что соответствует характерной ковалентной связи. Количество возможных связей ограничено также химической валентностью атома. Кластер можно масштабировать и вращать вокруг его геометрического центра. Программа позволяет пользователю создавать и удалять атомы, а также "перемещать" их с помощью компьютерной мыши. Таким образом, обеспечивается возможность конструирования новых конфигураций кластера. При запуске программы каждый атом определенного химического элемента имеет свой собственный цвет, заданный по умолчанию. Цвета каждого атома или группы атомов, а также их размеры, могут быть изменены (радиусы сфер и цилиндров по умолчанию равны 0.24 и 0.1 Å, соответственно). Также имеются инструменты для анализа геометрии кластера – вычисления длин связей и величин валентных углов.
Программа может работать в одном из двух режимов отображения – статическом или динамическом. В статическом режиме можно визуализировать кластеры, геометрическая конфигурация которых остается неизменной с течением времени, а динамический режим предоставляет инструментарий для работы с «фильмами». Каждый кадр такого «фильма» представляет собой некоторую новую геометрическую конфигурацию кластера. В динамическом режиме с каждым отдельным кадром можно работать точно так же, как и со статической конфигурацией: вращать, масштабировать, перемещать атомы, менять цвета и размеры, получать информацию длинах связей и валентных углах. Для работы в динамическом режиме предусмотрена специальная панель управления, которая позволяет пользователю запускать и останавливать воспроизведение фильма, сохранять или обрабатывать отдельные кадры, изменять скорость воспроизведения.
Программа может получать входные данные непосредственно из любой другой программы в виде четырех массивов, содержащих декартовы координаты атомов и их химические символы. Также возможно чтение входных данных из файлов (поддерживаются общепринятые форматы *.xyz и *.mol). При сохранении фильма все его кадры записываются подряд в один файл, а в первой строке указывается число атомов в кадре. Каждый отдельный кадр или весь фильм можно сохранять в этих форматах.
4. Краткий обзор пользовательского интерфейса
Программа имеет простой и интуитивно понятный интерфейс, управляемый при помощи компьютерной мыши. Все пространство окна программы разделено на три части – заголовок окна, строка меню и поле визуализации. На рис. 1 представлена визуализация молекулы брадикинина.
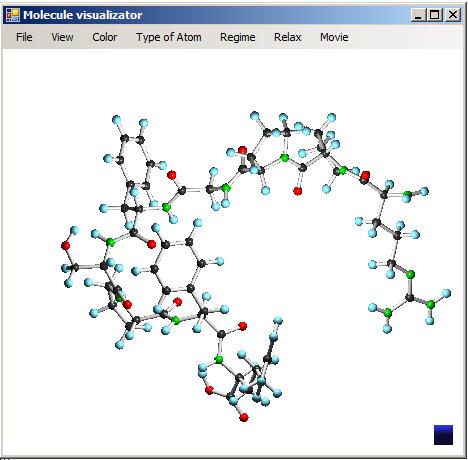
Рис. 1. Визуализация молекулярной структуры брадикинина.
В заголовке окна присутствуют три кнопки, типичные для операционной системы Windows, позволяющие свернуть, развернуть на весь экран и закрыть окно программы. Строка меню состоит из нескольких пунктов, важнейшие из которых – Файл, Вид, Тип атома, Режим и Релаксация. Меню «Файл» отвечает за работу с входными и выходными данными. Меню «Вид» позволяет установить удобный стиль отображения, а также открывать панель управления для динамического режима. Используя меню «Тип атома», пользователь может выбрать типы атомов, которые добавляются к кластеру при использовании молекулярного конструктора, вызываемого в меню "Режим".
Поле визуализации используется для отображения кластера и для управления процессом визуализации при помощи компьютерной мыши. Выделив мышью несколько атомов, можно определить длины связей и величины валентных углов, которые они образуют (см. рис. 2).
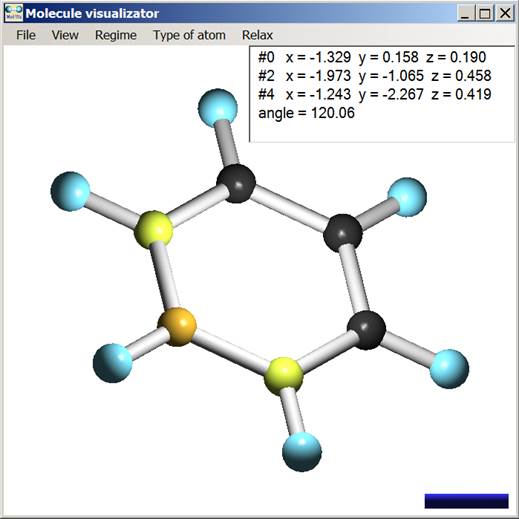
Рис. 2. Анализ молекулярной геометрии.
Для управления программой в динамическом режиме используется специальная панель менеджера фильмов, открывающаяся при переходе в динамический режим. Эта панель представлена на рис. 3 (на примере динамического изображения молекулы C6H12). Панель содержит стандартные элементы управления видео – набор кнопок для запуска и остановки воспроизведения, дорожку для прокрутки, дорожку для указания скорости воспроизведения, а также текстовое поле, в котором пользователь может указать номер нужного кадра.
Рис. 4 и рис. 5 иллюстрируют процесс оптимизации геометрии молекулы C4H10. Оптимизация основана на неортогональном алгоритме сильной связи [6], который был интегрирован с визуализатором (см. раздел 5).
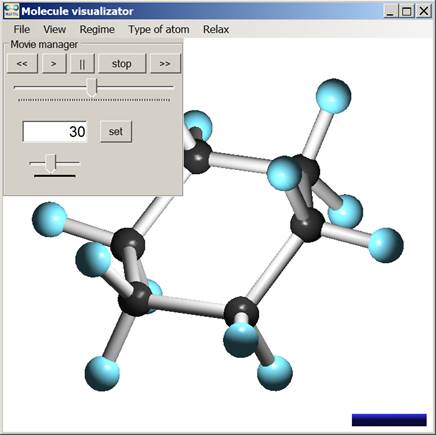
Рис. 3. Пользовательский интерфейс программы в динамическом режиме
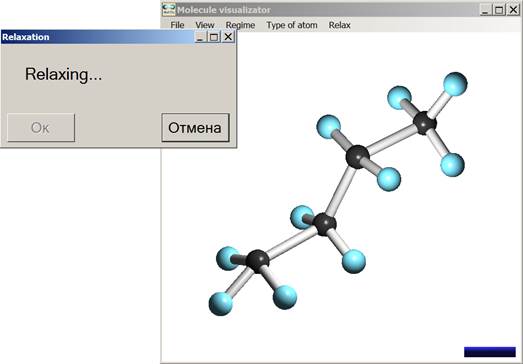
Рис. 4. Оптимизации геометрии молекулы C6H12
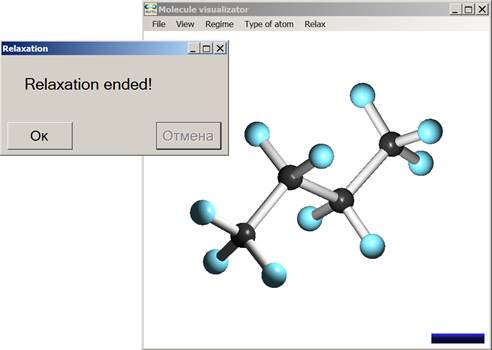
Рис. 5. Оптимальная геометрия молекулы C6H12
5. Интеграция визуализатора со сторонними программами
Чтобы продемонстрировать возможности нового визуализатора, мы использовали его для наглядного представления результатов работы программы, реализующей неортогональный алгоритм сильной связи [6]. Отметим, что программа была написана на языке Fortran, а визуализатор – на языке C#. Существует два распространенных способа, позволяющих организовать обмен данными между двумя программами, реализованными на разных языках программирования. Первый способ состоит в том, что программа визуализации вызывается из предварительно составленного исполняемого файла. При этом программы не могут выполняться параллельно, что приводит к снижению быстродействия и затрудняет обработку промежуточных данных.
Второй способ заключается в использовании технологии, основанной на динамических библиотеках. Этот подход требует модификации кода. Тем не менее, это позволяет организовать эффективное взаимодействие двух программ, контролируя процесс выполнение подпрограммы из связанной динамической библиотеки. Таким образом, появляется возможность остановить выполнение подпрограммы для изучения промежуточных результатов, которые могут быть сразу же отображены с использованием программы визуализации.
Учитывая перечисленные преимущества технологии динамических библиотек, мы использовали ее для объединения визуализатора с вычислительной программой, предназначенной для моделирования структуры кластеров в рамках модели сильной связи [6]. Чтобы апробировать получившийся программный комплекс, мы визуализировали процесс оптимизации геометрии около двухсот кластеров и молекул, в том числе колистина, брадикинина, графена и поликубанов. Все эти соединения были успешно визуализированы при помощи представляемой программы.
ЗАКЛЮЧЕНИЕ
Представлена новая программа для визуализации атомных кластеров и молекулярных структур. Она может использоваться для обработки выходных файлов сторонних программ, реализующих моделирование атомных кластеров, или внедряться в такие программы в качестве динамически подключаемой библиотеки. Это обеспечивает совместное использование оперативной памяти, что увеличивает производительность системы и позволяет «на лету» визуализировать получаемые данные. Мы надеемся, что новый визуализатор будет полезен для исследователей, работающих в области нанофизики, химии и биологии.
БЛАГОДАРНОСТИ
Работа поддержана в рамках Федеральной Целевой Программы «Исследования и разработки по приоритетным направлениям развития научно-технологического комплекса России на 2014-2020 годы», соглашение № 14.578.21.0064.
Используемая литература
1 http://www.msg.chem.iastate.edu/gamess/index.html Accepted date: 15.12.2014.
2 http://www.yaehmop.sourceforge.net Accepted date: 15.12.2014
3 http://www.chemcraftprog.com/ Accepted date: 15.12.2014.
4 http://avogadro.cc/wiki/Main_Page Accepted date: 15.12.2014.
5 http://www.wavefun.com/ Accepted date: 15.12.2014.
6 K.P. Katin, M.M. Maslov, Thermal stability of nitro derivatives of hydrocarbon cubane // Russ. J. Phys. Chem. B, Vol. 5, pp 770-779, 2011.
7 D. D. Holm, Geometric Mechanics, Part II: Rotating, Translating and Rolling / Mathematics Department Imperial College London, 2013.
8 H. Goldstein, Classical Mechanics, 2nd edition / Reading, MA: Addison-Wesley, 1980.
9 E. W. Swokowski, Calculus with Analytic Geometry (The Prindle, Weber & Schmidt series in mathematics), 4th edition / Pws Pub Co, 1988.
10 K. Shoemake, Animating Rotation with Quaternion Curves // Computer Graphics, Vol. 19, pp 245-254, 1985.
11 H. Schildt, C# 4.0. The Complete Reference / McGraw Hill, 2010.
12 R. S. Wright Jr., B. Lipchak, OpenGL Super Bible, 3rd edition / Sams, 2004.
CLUSTVIS1: A NEW SOFTWARE PACKAGE FOR VISUALIZATION OF ATOMIC CLUSTERS AND MOLECULES
A.Yu. Pischulina1, S.A. Shostachenko, K.P. Katin, V.S. Prudkovskiy2,3, M.M. Maslov, R.V. Ryzhuk, N.I. Kargin
National Research Nuclear University MEPhI, Moscow, 115409 Russia
1 All-Russian Research Institute for Nuclear Power Plants Operation, Moscow, 109507 Russia
2 CNRS, LAAS, 7 avenue du colonel Roche, F-31400 Toulouse, France
3 University de Toulouse, LAAS, F-31400 Toulouse, France
A new software package for visualization of atomic clusters and molecules is designed. Instead of analogous software products, this one compiled as a dynamic link library. It allows easy integration with any modeling program and “on-fly” visualization of calculated results. The software provides static and dynamic clusters and molecules visualization in atom-bond notation. Carried out tests prove the ability of new package to visualize a wide range of atomic clusters and nanostructures, included large biological macromolecules.
Keywords: nanostructural modeling, molecular dynamics, structural relaxation, atomic animation.
REFERENCES
1 http://www.msg.chem.iastate.edu/gamess/index.html Accepted date: 25.12.2014.
2 http://www.yaehmop.sourceforge.net Accepted date: 25.12.2014.
3 http://www.chemcraftprog.com/ Accepted date: 25.12.2014.
4 http://avogadro.cc/wiki/Main_Page Accepted date: 25.12.2014.
5 http://www.wavefun.com/ Accepted date: 25.12.2014.
6 K.P. Katin, M.M. Maslov, Thermal stability of nitro derivatives of hydrocarbon cubane // Russ. J. Phys. Chem. B, Vol. 5, pp 770-779, 2011.
7 D. D. Holm, Geometric Mechanics, Part II: Rotating, Translating and Rolling / Mathematics Department Imperial College London, 2013.
8 H. Goldstein , Classical Mechanics, 2nd edition / Reading, MA: Addison-Wesley, 1980.
9 E. W. Swokowski, Calculus with Analytic Geometry (The Prindle, Weber & Schmidt series in mathematics), 4th edition / Pws Pub Co, 1988.
10 K. Shoemake, Animating Rotation with Quaternion Curves // Computer Graphics, Vol. 19, pp 245-254, 1985.
11 H. Schildt, C# 4.0 The Complete Reference / McGraw Hill, 2010.
12 R. S. Wright Jr., B. Lipchak, OpenGL Super Bible, 3rd edition / Sams, 2004.